Košice
Ve dnech 21. - 23. 9. 2022 jsme se aktivně zúčastnili XXXII. Izakovičova Memoriálu v Košicích pořádaný společností lékařské genetiky. SPADIA získala ocenění za nejlepší poster.

Ve dnech 21. - 23. 9. 2022 jsme se aktivně zúčastnili XXXII. Izakovičova Memoriálu v Košicích pořádaný společností lékařské genetiky. SPADIA získala ocenění za nejlepší poster.
Naši kolegové z oddělení molekulární biologie se aktivně zúčastnili jednoho z nejdůležitějších kongresů v oblasti genetiky na Slovensku, kde přednesli své zkušenosti prostřednictvím dvou přednášek a dvou posterů.
MUDr. Marek Godava, Ph.D.. přednesl dvě přednášky:
Od mozaiky k delécii – fenotypové spektrum pacientov s mutáciou v PAX6
Vrodená anirídia je pomerne vzácne ochorenie, uvádzaná incidencia je približne 1/65 000 – 96 000. I keď ide o charakteristický a často dominujúci rys ochorenia, tak postupne dochádza k postihnutiu aj ďalších štruktúr oka. Ide o panokulárne postihnutie s dominujúcou dysgenézou predného segmentu oka. Ochorenie je spôsobené poruchou vývoja v dôsledku mutácie v PAX6, dedičný prenos je autozómovo dominantný. Významnú variabilitu fenotypu prezentujeme v súbore 13 prípadov (sporadických aj familiárnych) s výskytom mutácií bodových, 1x v mozaike a 1x v dôsledku veľkej delécie.
Xeroderma pigmentosum s dominujúcim neurologickým postihnutím
Prezentujeme 44-ročného neurologického pacienta, ktorý sa sťažuje hlavne na kŕče končatín, a to od ranej mladosti. V neurologickom náleze bola opisovaná neuromyotónia (uvažovalo sa o Isaacovom syndróme), oro-facio-skapulárne dyskinézie, choreoatetózny syndróm, kvadruataxia, neskôr i fascikulácie. Takisto je zvýšená fotosenzitivita a prítomné poikilodermie. V 43 rokoch bola zistená mentálna porucha pri výraznej atrofii mozgu, je prítomná aj axonálne demyelinizačná neuropatia dolných končatín. Už vo veku 29 rokov bola vylúčená Huntingtonova chorea, VLCFA v sére bolo v norme, uvažovalo sa o miernej forme xeroderma pigmentosum (XP). Celoexómové sekvenovanie odhalilo mutáciu c.772_785del v XPA v homozygotnej forme. Táto mutácia už bola opísaná v zloženej heterozygotnej forme v rodine s miernou formou XP, kde taktiež dominovali neurologické ťažkosti.
Mgr. Václav Palata, prezentoval svou práci prostřednictví posteru, který byl zvolen jako nejlepší poster konference:
Molekulárně-genetická analýza genů spojených s hereditárními nádorovými syndromy
Hereditární nádorové syndromy jsou malou, ale z klinického hlediska velice významnou skupinou onkologických onemocnění. Vznik nádorového onemocnění je komplexní a polyfaktoriální proces. Pouze 5 – 10 % všech nádorových onemocnění může mít dědičnou formu. Pro tyto nádorové syndromy je typický častý výskyt konkrétních typů nádorů, mnohočetných malignit a nádorů v rodině mezi blízce příbuznými jedinci. Za jejich vznik je zodpovědná germinální mutace zděděná od jednoho či obou rodičů, která může být zároveň i jakýmsi spouštěčem nekontrolovaného nádorového bujení, jelikož často postihuje geny regulující buněčnou proliferaci a opravu poškozené DNA. Penetrance, neboli pravděpodobnost s jakou se daná zárodečná mutace projeví ve fenotypu jedince, je značně vysoká a dosahuje mnohonásobku populačního rizika onemocnění. Nádorové syndromy jsou zpravidla velmi heterogenní skupinou onemocnění rozdílné jak ve svém genotypu, tak i svými fenotypovými projevy. Například v rámci jedné rodiny mohou mít nosiči stejné mutace rozdílný klinický projev onemocnění a naopak, výskyt různých mutací může vést k fenotypovému projevu konkrétního syndromu. Ve většině případů se vyskytuje jedna patogenní mutace v jednom konkrétním genu. Výskyt dvou patogenních mutací je poměrně vzácný jev. Výjimka ale potvrzuje pravidlo. V jedné námi vyšetřované rodině se vyskytují společně dvě patogenní mutace, a to v genech BRCA2 a MUTYH.
Mgr. David Kaspřák, Ph.D. představil poster na téma:
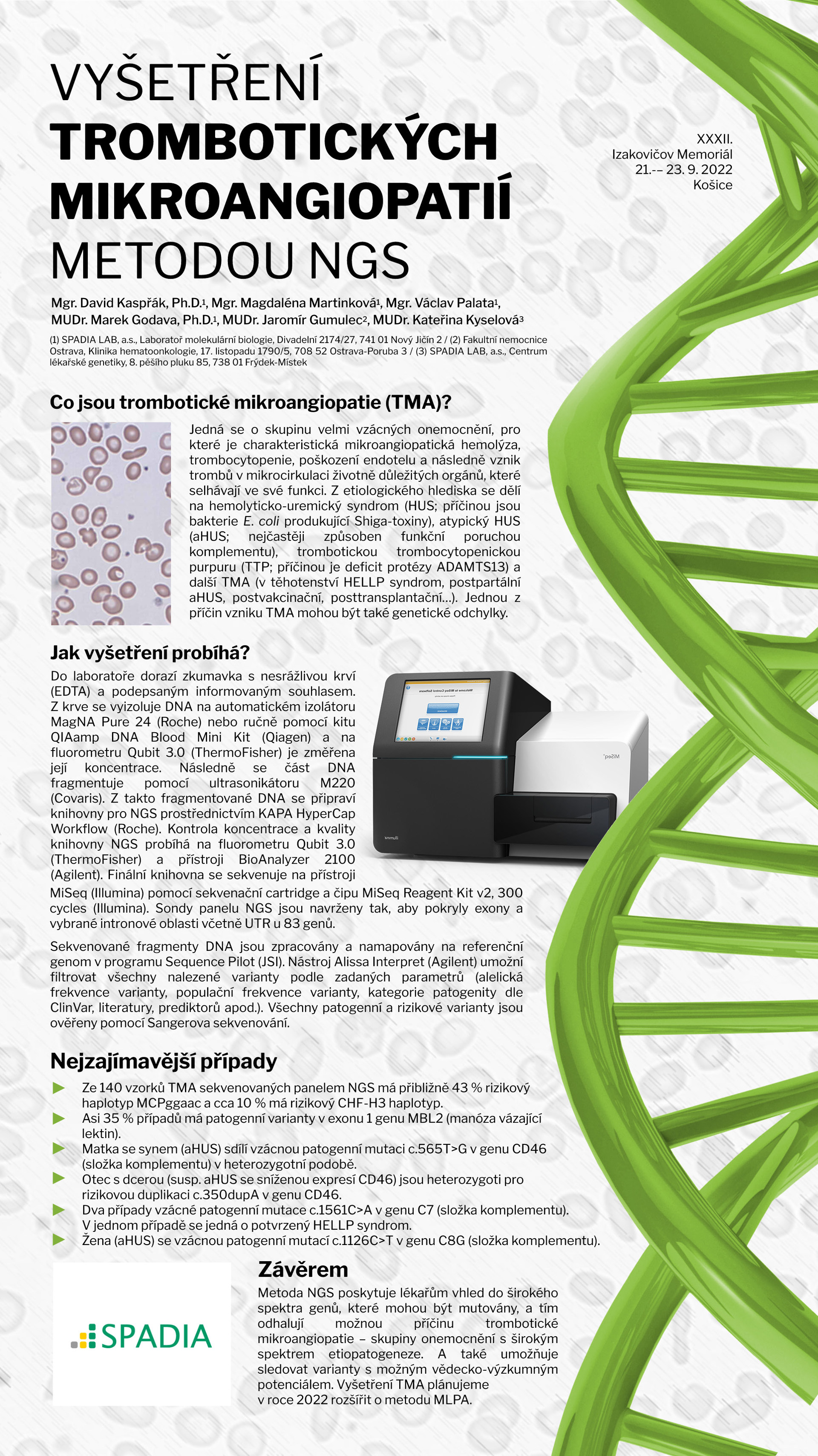
Vyšetření trombotických mikroangiopatií metodou NGS
Trombotické mikroangiopatie (TMA) jsou skupina velmi vzácných onemocnění, pro kterou je charakteristická mikroangiopatická hemolýza, trombocytopenie, poškození endotelu a následně vznik trombů v mikrocirkulaci životně důležitých orgánů. Jednou z příčin TMA mohou být genetické odchylky, které nacházíme u asi 50 – 60 % pacientů s TMA. Závažné genetické mutace nalézáme nejčastěji v genech pro komplementový systém. Mezi TMA ale spadá i řada patologických stavů, jejíž etiopatogeneze je dosti široká, a ne vždy musí nutně souviset s komplementem. Z těchto důvodů byl sestaven panel genů pro vyšetření široké škály komplementopatií a také pro analýzu genů, které nesouvisí nebo nepřímo souvisí s komplementovým systémem, ale jejich mutace mohou zapříčinit rozvoj TMA. Mutační analýza je prováděná na oddělení molekulární biologie SPADIA LAB od roku 2018 metodou Next Generation Sequencing (NGS) a cílem tohoto příspěvku je představit podrobnosti tohoto vyšetření.